







Genes 2025, 16(5), 567; https://doi.org/10.3390/genes16050567 (registering DOI) - 10 May 2025
Abstract
Background/Aims: Ledrinae comprises about 460 described species across five tribes and represents an early-branching, morphologically distinctive lineage of leafhoppers, yet its intra-subfamilial relationships remain ambiguous owing to limited mitogenomic sampling. Here, we sequence and annotate the complete mitochondrial genome of Petalocephala arcuata—only
[...] Read more.
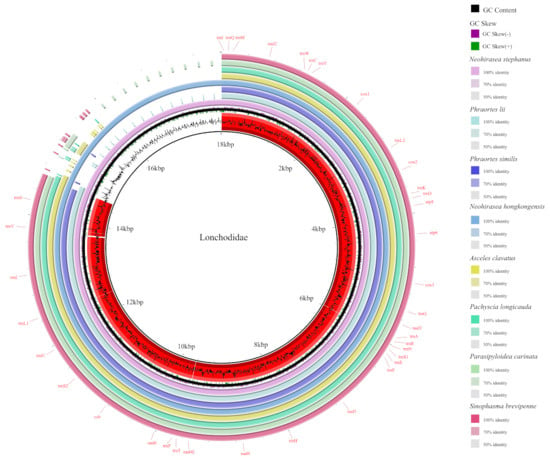
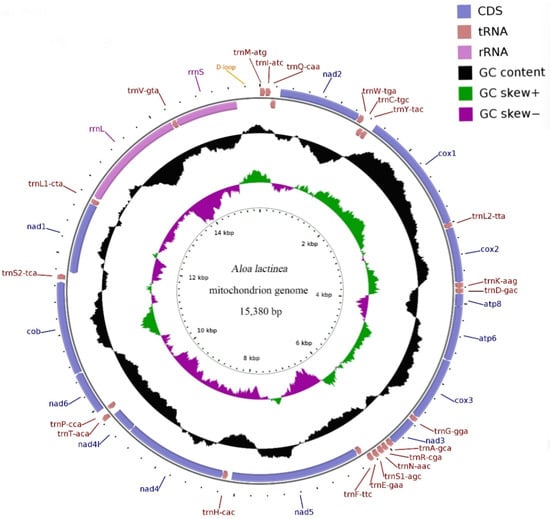
Background/Aims: Ledrinae comprises about 460 described species across five tribes and represents an early-branching, morphologically distinctive lineage of leafhoppers, yet its intra-subfamilial relationships remain ambiguous owing to limited mitogenomic sampling. Here, we sequence and annotate the complete mitochondrial genome of Petalocephala arcuata—only the 18th Ledrinae mitogenome—to broaden taxon coverage within the genus and furnish critical molecular data for rigorously testing Ledrinae monophyly and refining tribal and genus level phylogenetic hypotheses. Methods: In this study, we sequenced and annotated the complete mitochondrial genome of P. arcuata via Illumina sequencing and de novo assembly, and reconstructed the phylogeny of 62 Cicadellidae species using maximum likelihood and Bayesian inference methods. Results: The 14,491 bp circular mitogenome of P. arcuata contains 37 genes with 77.4% A+T. All PCGs use ATN start codons except ND5 (TTG), and codon usage is A or U biased. Of 22 tRNAs, only trnS1 lacks a DHU arm, while the others adopt the canonical cloverleaf structure. Bayesian inference and maximum likelihood analyses produced broadly congruent topologies with mostly high nodal support, recovering Ledrinae as monophyletic and clustering all Petalocephala species into a well‑supported clade. Conclusions: In this study, we enriched the molecular resources for the genus Petalocephala by sequencing, annotating, and analyzing the complete mitochondrial genome of P. arcuata. Phylogenetic reconstructions based on these genomic data align closely with previous morphological diagnoses, further confirming the monophyly of the genus Petalocephala.
Full article
(This article belongs to the Special Issue Molecular Evolution, Mitochondrial Genomics and Mitochondrial Genome Expression in Animals: 2024–2025)







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}